
Why protein aggregation hides for years


One of the central questions in neurodegenerative disease revolves around timing. Proteins such as amyloid-β, tau, and α-synuclein are always present inside of our cells, yet for most of our lives they remain stable and perform their normal functions. For some people, usually later in life, these proteins can start to accumulate and form aggregates which are linked to all sorts of diseases. How can a cell maintain this balance for so long, and suddenly crash into aggregation?
A few years ago I went to the chaperone meeting in the Netherlands, where Harm Kampinga presented his vision of why this happens. In his view (simplified and by my memory), there is a balance between cells accumulating damage and repairing that damage. Accumulation of damage is simple for stress, proteins that randomly misfold, mutations, all kinds of things you cannot avoid when systems age. At the same time, these problems get repaired by chaperones and other systems (also called proteostasis), which keeps the cell running healthily. This system also accumulates damage over time, and repair gets less efficient. At some point, you may cross over a boundary where cells can no longer repair the damage, and we enter the area of rampant aggregation. This can be due to worse repair, or more and more damage accumulating (including "peaks" in the damage, such as infections, physical trauma, etc.). In my opinion, this is quite a nice high-level view of thinking about why systems get thrown out of balance. The article I'm discussing today takes a more mathematical approach to understanding what is happening.
In recent work by Cotton et al. (Meisl group) in The Journal of Chemical Physics, they describe how the balance of aggregate formation and cell clearance can give bistability - a system in which we end up in one of two states. In this paper they view the cell in terms of monomeric protein concentration and aggregate load, then ask which combinations are stable and which ones lead to runaway growth.
When they first analyze the simplest case, where clearance scales proportionally with the amount of aggregate, the result is already informative. There is a critical monomer concentration below which the system approaches a finite steady state, and above which aggregate mass grows without bound. This suggests that relatively modest changes in monomer levels can move a system across a threshold, meaning that small changes could lead to onset of disease. The more interesting result is when they make clearance more realistic (biologically, machinery like lysosomes or proteasomes get overwhelmed at some point), and allow it to saturate. Once removal has a maximum capacity, the system has bistability. Cells can remain in a low-aggregate state for long periods, yet once aggregate burden crosses a boundary, clearance can no longer keep up and the dynamics run away. As the authors describe, it's like a tug-of-war between clearance of protein and pressure, or as I would, a tub that slowly fills up but also has a drain at the bottom. At some point the tub overflows, or the tug-of-war is won.
Instead of just looking at reaction rates equations, they map the system to a plane based on monomer concentration and aggregate mass, called a phase plane. In this representation, health and pathology (in this case, just described as protein aggregation) are different regions of the landscape. If you increase the monomer concentration or suddenly add seeds, the system can enter a regime where aggregation reinforces itself. This provides a concise mathematical explanation for two frequently discussed observations: the effectiveness of seeding and the potential of monomer-lowering interventions to restore stability.
Next they vary the removal method, the aggregation mechanism, and even consider cases with finite carrying capacity or conserved total protein. In these variants of the main system they find a tipping-point and bistability. The advantage of this kind of modeling is that even if we don't know the exact biological numbers and variables, the theory is robust enough that there's likely some truth to it.
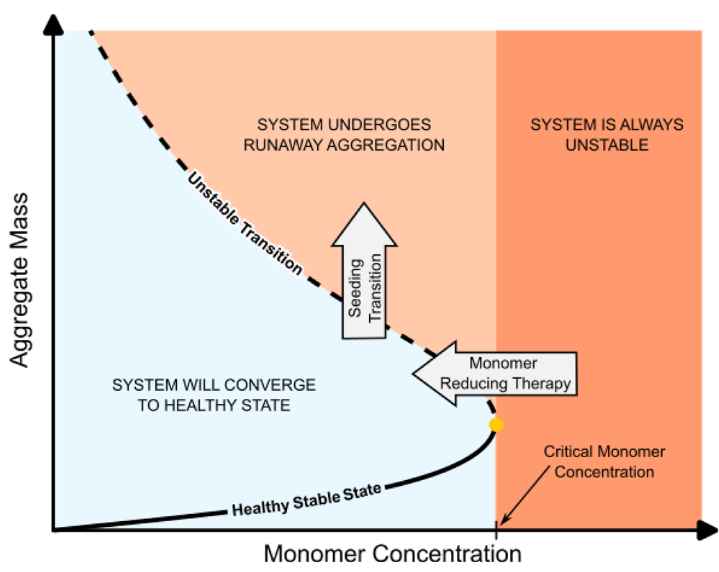
What this opens up, of course, are experimental questions. If this phase plane is the right high-level picture, where do real cells sit on it? How close are healthy neurons to the boundary, and what can we do to help them stay on the right side? Does aging mainly push systems by increasing the effective damage load, by weakening clearance, by raising monomer levels, or by nudging several of these at once? And how should we think about transient shocks such as inflammation, infection, or trauma, which might briefly move a system across a threshold even if it was previously stable?
Overall, this is impressive and useful work in my opinion. It takes a complicated molecular problem and extracts a mathematically clean structure from it, while still preserving the relevant biological details. Papers like this give us a map of possibility, which we can hopefully use to navigate our experimental world.



